



Wissenschaftler haben ein Enzym identifiziert, das möglicherweise eine entscheidende Rolle bei der Huntington-Krankheit spielt, einer seltenen und tödlichen Krankheit, die zur Zerstörung von Gehirnzellen führt. Forscher sagen, dass die Entdeckung eines wichtigen Enzyms, das an der Huntington-Krankheit beteiligt ist, den Weg für zukünftige Behandlungen zur Vorbeugung der Krankheit ebnen könnte.
Neue Forschungen an Nagetieren und Menschen haben gezeigt, dass der Spiegel eines bestimmten Enzyms, der Glutathion-S-Transferase Omega-2 (GSTO2), im Gehirn ansteigt, noch bevor Symptome der Huntington-Krankheit auftreten.
Diese am 28. Oktober 2024 in der Fachzeitschrift Nature Metabolism veröffentlichten Ergebnisse könnten auf neue Wege zur Vorbeugung der Huntington-Krankheit hinweisen, bevor sie sich entwickelt, sagen die Autoren der Studie. Zukünftige Behandlungen könnten Medikamente umfassen, die GSTO2 blockieren, um das Fortschreiten der Krankheit zu stoppen oder zu verlangsamen.
Die Huntington-Krankheit ist eine Erbkrankheit, die durch eine Mutation im HTT-Gen verursacht wird, das Anweisungen für ein Protein namens Huntingtin enthält. Ein Elternteil, der dieses mutierte Gen trägt, hat eine 50-prozentige Chance, die Huntington-Krankheit an jedes Kind weiterzugeben.
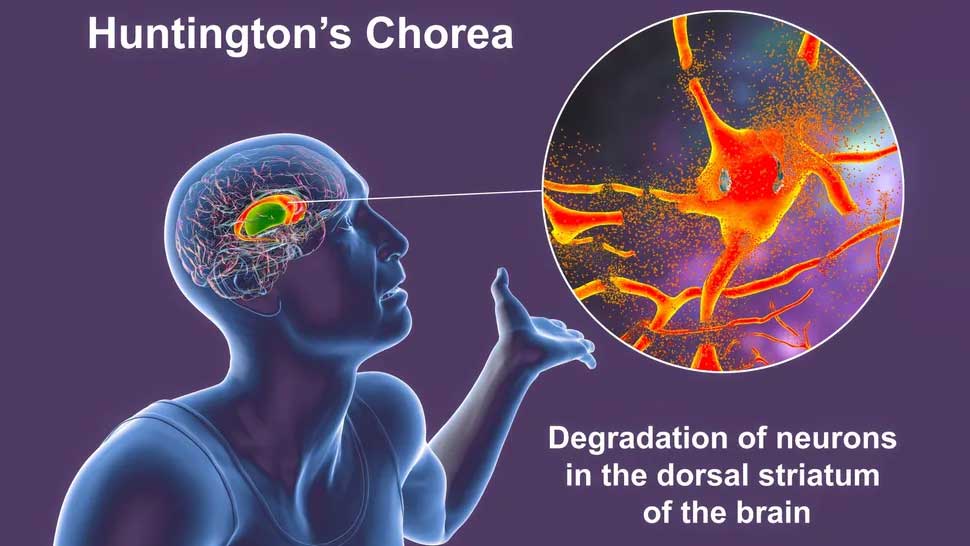
Die Mutation führt dazu, dass Zellen zu viel Dopamin produzieren – einen wichtigen chemischen Botenstoff im Gehirn – und dies führt zum Abbau bestimmter Neuronen. Dieser Zusammenbruch ist in einem Teil des Gehirns, dem Striatum, besonders schwerwiegend und führt dazu, dass die Patienten kognitive und motorische Symptome entwickeln. Dazu können Schwierigkeiten beim Gehen, unwillkürliches Zucken und Konzentrationsschwierigkeiten gehören.
Die Symptome der Huntington-Krankheit treten normalerweise im Alter zwischen 30 und 50 Jahren auf. Dieser Zustand beeinträchtigt allmählich die Funktionsfähigkeit des Patienten und führt schließlich etwa 10 bis 30 Jahre nach Auftreten der Symptome zum Tod.
Bisher konnten Wissenschaftler nicht erklären, warum die HTT-Mutation zu einer übermäßigen Dopaminproduktion führt. Dies ist einer der Gründe, warum es keine Heilung für die Huntington-Krankheit gibt – die verfügbaren Medikamente helfen nur, die Symptome zu lindern, nachdem der Schaden bereits angerichtet ist. Das HTT-Gen ist auch im gesamten Körper aktiv, was die Entwicklung gezielter Behandlungen zur Bekämpfung seiner Auswirkungen auf das Gehirn erschwert.
In der neuen Studie verfolgten die Forscher einen anderen Ansatz: „Anstatt die Mutation in diesem bestimmten Gen zu untersuchen, die die Huntington-Krankheit verursacht, haben wir uns die Signale angesehen, die diese Mutation beeinflusst, und was sie bewirken“, sagte Moderatorin Liliana Minichiello gegenüber Live Wissenschaftlicher Autor der Studie und Professor für Zelluläre und Molekulare Neurowissenschaften an der Universität Oxford.
Gehirnzellen geben einander Chemikalien weiter, um Signale zu übertragen. Darüber hinaus helfen Kettenreaktionen chemischer Aktivität in jeder Gehirnzelle den Neuronen, zu überleben, zu wachsen und ihre Integrität aufrechtzuerhalten. Es gibt beispielsweise Signale, die benötigt werden, um Neuronen am Leben zu halten, und diese Signale sind bekanntermaßen bei der Huntington-Krankheit gestört. Bestimmte Neuronen im Striatum sind am anfälligsten dafür, dass ihre Signalübertragung auf diese Weise gestört wird.
Um diese Idee weiter zu untersuchen, züchteten die Forscher gentechnisch veränderte Mäuse, deren Striatalzellen nicht in der Lage waren, diese wichtigen Überlebenssignale zu erzeugen. Sie stellten fest, dass der Dopaminspiegel im Gehirn der Nagetiere mehrere Monate anstieg, bevor die Nagetiere irgendwelche motorischen Symptome zeigten, die einer frühen Form der Huntington-Krankheit ähnelten.
Durch die Messung der Genaktivität striataler Neuronen fanden die Forscher heraus, dass die Störung der Zellüberlebenssignale offenbar die Menge an GSTO2 in den Zellen erhöht. Letztendlich führte dieser Anstieg des GSTO2 zu einer erhöhten Dopaminproduktion und einer fortschreitenden motorischen Dysfunktion bei Mäusen.
Das Team stellte fest, dass die Blockierung von GSTO2 diesen gesamten Prozess verhinderte.
In separaten Experimenten beobachteten die Forscher einen ähnlichen Anstieg von GSTO2 im Gehirn von Ratten mit einer der Huntington-Krankheit ähnlichen Erkrankung sowie im Gehirngewebe von Patienten mit der Huntington-Krankheit. Sowohl Ratten als auch Menschen zeigten diesen Anstieg des GSTO2, entwickelten jedoch noch keine erkennbaren Symptome dieser Erkrankungen.
Zusammengenommen verdeutlichen diese Ergebnisse spezifische zelluläre Veränderungen, die den Ausbruch der Huntington-Krankheit auslösen können.
Forscher müssen sich nun die Rolle von GSTO2 bei Nagetieren, die die HTT-Mutation tragen, genauer ansehen, um weiter zu testen, ob der Zusammenhang kausal ist. Wenn ja, könnte GSTO2 ein potenzielles neues Ziel für Medikamente sein, die das Fortschreiten der Huntington-Krankheit stoppen oder verlangsamen sollen, schlug Minichiello vor.
