



Les scientifiques ont identifié une enzyme qui pourrait jouer un rôle crucial dans la maladie de Huntington, une maladie rare et mortelle qui provoque la destruction des cellules cérébrales. Les chercheurs affirment que la découverte d’une enzyme importante impliquée dans la maladie de Huntington pourrait ouvrir la voie à de futurs traitements visant à prévenir la maladie.
De nouvelles recherches chez les rongeurs et les humains ont montré que les niveaux d’une enzyme spécifique, la glutathion S-transférase oméga-2 (GSTO2), augmentent dans le cerveau avant même l’apparition des symptômes de la maladie de Huntington.
Ces résultats, publiés le 28 octobre 2024 dans la revue Nature Metabolism, pourraient ouvrir la voie à de nouvelles façons de prévenir la maladie de Huntington avant qu’elle ne se développe, affirment les auteurs de l’étude. Les futurs traitements pourraient inclure des médicaments qui bloquent GSTO2 pour arrêter ou ralentir la progression de la maladie.
La maladie de Huntington est une maladie héréditaire causée par une mutation du gène HTT, qui transporte les instructions d’une protéine appelée huntingtine. Un parent porteur de ce gène mutant a 50% de chances de transmettre la maladie de Huntington à chaque enfant.
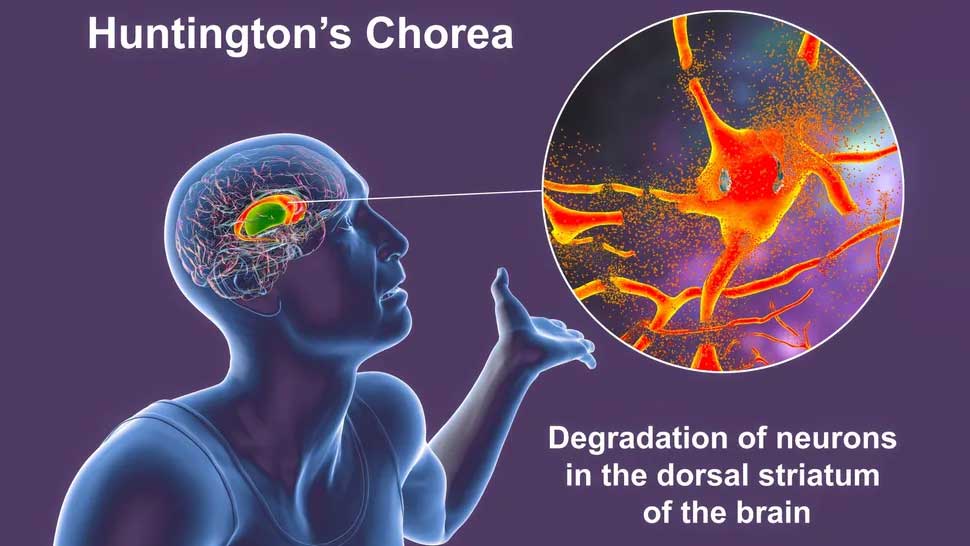
La mutation amène les cellules à produire trop de dopamine – un messager chimique clé dans le cerveau – et cela conduit à la dégradation de certains neurones. Cette dégradation est particulièrement grave dans une partie du cerveau appelée striatum, provoquant le développement de symptômes cognitifs et moteurs chez les patients. Ceux-ci peuvent inclure des difficultés à marcher, des secousses involontaires et des difficultés à se concentrer.
Les symptômes de la maladie de Huntington commencent généralement à apparaître entre 30 et 50 ans. Cette condition altère progressivement la capacité fonctionnelle du patient, conduisant finalement à la mort environ 10 à 30 ans après l’apparition des symptômes.
Jusqu’à présent, les scientifiques ne parvenaient pas à expliquer pourquoi la mutation HTT entraîne une production excessive de dopamine. C’est l’une des raisons pour lesquelles il n’existe aucun remède contre la maladie de Huntington: les médicaments disponibles n’aident à soulager les symptômes qu’une fois les dégâts déjà causés. Le gène HTT est également actif dans tout le corps, ce qui rend difficile le développement de traitements ciblés qui combattent ses effets sur le cerveau.
Dans la nouvelle étude, les chercheurs ont adopté une approche différente: «Au lieu d’examiner la mutation de ce gène particulier qui cause la maladie de Huntington, nous avons examiné les signaux que cette mutation affecte et ce qu’ils font», a déclaré Liliana Minichiello, présentatrice, à Live. Auteur scientifique de l’étude et professeur de neurosciences cellulaires et moléculaires à l’Université d’Oxford.
Les cellules cérébrales se transmettent des produits chimiques pour transmettre des signaux. De plus, au sein de chaque cellule cérébrale, les réactions en chaîne de l’activité chimique aident les neurones à survivre, à se développer et à maintenir leur intégrité. Par exemple, certains signaux sont nécessaires pour maintenir les neurones en vie, et on sait que ces signaux sont perturbés dans la maladie de Huntington. Certains neurones du striatum sont les plus vulnérables à une telle perturbation de leur signalisation.
Pour approfondir cette idée, les chercheurs ont élevé des souris génétiquement modifiées dont les cellules striatales étaient incapables de produire ces signaux clés de survie. Ils ont remarqué que les niveaux de dopamine dans le cerveau des rongeurs augmentaient plusieurs mois avant que les rongeurs ne présentent des symptômes moteurs ressemblant à une forme précoce de la maladie de Huntington.
En mesurant l’activité génétique des neurones striataux, les chercheurs ont découvert que la perturbation des signaux de survie cellulaire semblait augmenter la quantité de GSTO2 dans les cellules. En fin de compte, cette augmentation de GSTO2 a entraîné une augmentation de la production de dopamine et un dysfonctionnement moteur progressif chez la souris.
L’équipe a constaté que le blocage de GSTO2 empêchait tout ce processus.
Dans des expériences distinctes, les chercheurs ont observé une augmentation similaire de GSTO2 dans le cerveau de rats atteints d’une maladie similaire à la maladie de Huntington, ainsi que dans le tissu cérébral de patients atteints de la maladie de Huntington. Les rats et les humains ont montré cette augmentation de GSTO2, mais n’ont pas encore développé de symptômes notables de ces conditions.
Pris ensemble, ces résultats mettent en évidence des changements cellulaires spécifiques susceptibles de déclencher l’apparition de la maladie de Huntington.
Les chercheurs doivent maintenant examiner de plus près le rôle de GSTO2 chez les rongeurs porteurs de la mutation HTT afin de déterminer si le lien est causal. Si tel est le cas, GSTO2 pourrait être une nouvelle cible potentielle pour les médicaments conçus pour arrêter ou ralentir la progression de la maladie de Huntington, a suggéré Minichiello.
